FSL系列教程 #5. FSL脚本编写
简介
在对单个受试者的单次run进行预处理并建立模型之后,我们就需要对数据集中所有受试者的所有run进行相同的处理。这看似繁琐,但也是可行的–我们只有26个受试者,每个受试者有两次run。我们可能认为这可以在一周左右的时间内完成;而且也可以随时将这项任务分配给几名研究助理。
然而,在某些时候,您会遇到两个问题:
- 我们会发现,手动分析每次运行不仅乏味,而且容易出错,随着要分析的运行数量增加,出错的概率也会显著增加;
- 对于较大的数据集(例如,80个受试者,每个受试者有5次run),这种方法很快就会变得不切实际。
另一种方法是编写分析脚本。就像演员有一个脚本,告诉他说什么、站在哪里、在哪里移动一样,我们也可以编写一个脚本,告诉计算机如何分析数据集。这样做有双重好处,既可以实现分析自动化,又可以分析任何规模的数据集–分析两个受试者或两百个受试者的代码几乎完全相同。
首先,我们将创建一个模板,其中包含分析单次run所需的代码,然后我们将使用for-loop自动分析所有运行。这个想法很简单;尽管代码一开始可能难以理解,但一旦我们对其更加熟悉,我们就会发现如何将其应用于任何数据集。
理解下述代码脚本需要Linux的shell脚本基础,推荐掌握了基础的shell脚本语法后,再来继续阅读下述内容。
创建脚本模版
当分析sub-08被试的第一个run时,会创建一个名为run1.feat的目录。在该目录下有几个文件和子目录。其中一个文件design.fsf包含了从FEAT图形用户界面转录到文本文件中的所有代码。这是FSL运行来完成每个预处理和建模步骤的代码。如果在文本编辑器中打开design.fsff文件和用来创建design.fsf文件的FEAT图形用户界面并排比较,我们可以看到输入到FEAT图形用户界面的数据在哪里被写入design.fsf文件。
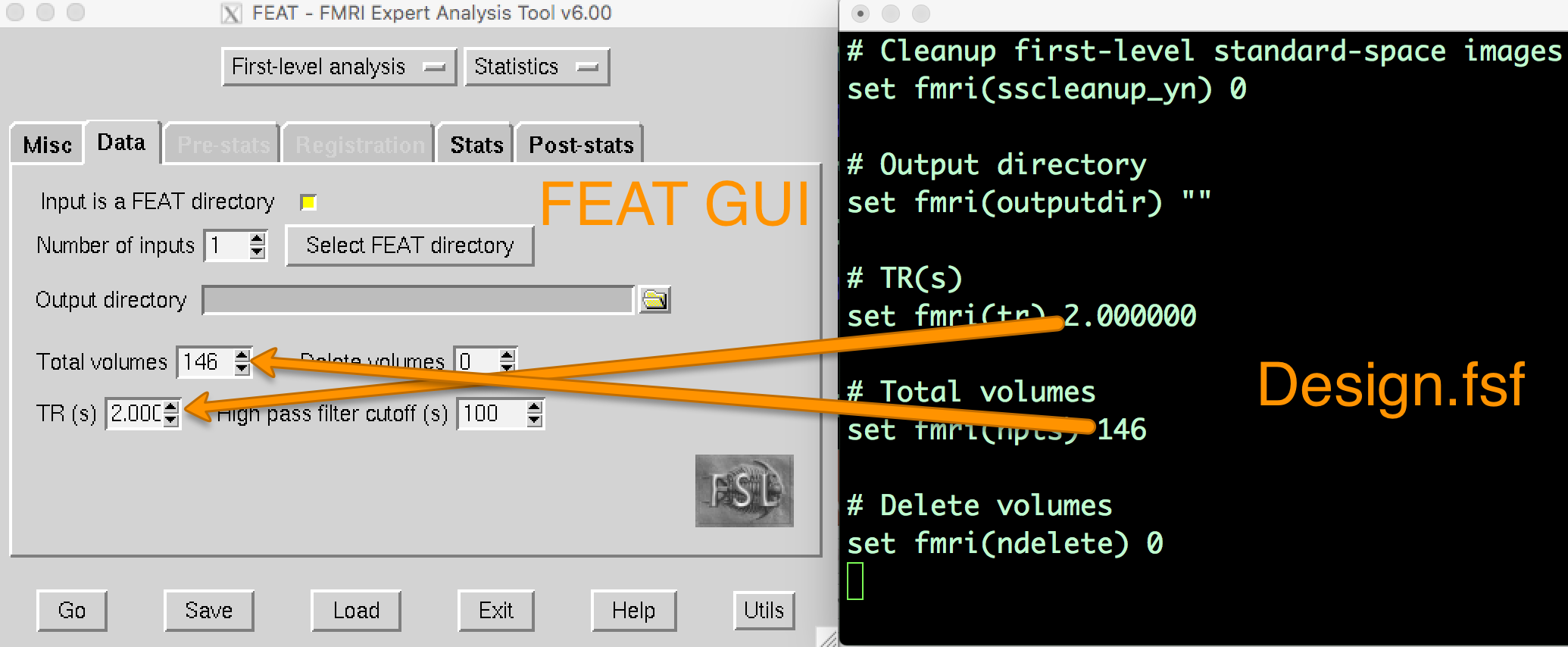
如果我们打开了FEAT图形用户界面,点击屏幕底部的Load按钮,并选择run1.feat目录下的design.fsf文件,它将把所有设置更改为您保存脚本时在图形用户界面中输入的设置。
在前面的教程中,我们分别运行了FEAT的预处理和模型拟合。现在我们将从FEAT GUI的下拉菜单中选择 “Full Analysis(全分析)",创建一个将这两个步骤结合在一起的模板。
首先,输入rm -r run1.feat删除当前run1.feat目录。然后在命令行输入Feat_gui打开FEAT图形用户界面。我们将在下拉菜单中选择 “Full Analysis(全面分析)“选项,而不是将其作为两个单独的会话来运行。以前面的教程为指导,填写预处理和模型拟合的所有必填项。
填写完所有字段后,不要点击Go按钮,而是点击Save并标注文件为design_run1。这将保存多个扩展名为 “con”、“mat “和 “png “的文件,但我们的脚本将使用文件design_run1.fsf。
现在对run 2执行相同的步骤,加载相应的功能数据和时序文件。将文件保存为design_run2.fsf。
在代码编辑器(或者文本编辑器)中打开design_run1.fsf,查看所有已填写的选项。我们的目标是使该模板可用于任何被试,只需在for-loop中稍作改动即可。在本例中,我们唯一需要更改的是被试名称,其余选项对每个被试都是相同的。
运行脚本
将design_run1.fsf和design_run2.fsf文件移到包含被试的目录下(即mv design*.fsf ..,然后cd ..)。然后下载脚本 run_1stLevel_Analysis.sh(如下述代码所示),并将其移动到Flanker目录中:
#!/bin/bash
# Generate the subject list to make modifying this script
# to run just a subset of subjects easier.
# 依次遍历26个被试数据
for id in `seq -w 1 26` ; do
subj="sub-$id"
echo "===> Starting processing of $subj"
echo
cd $subj
# 颅骨去除
# If the brain mask doesn’t exist, create it
if [ ! -f anat/${subj}_T1w_brain_f02.nii.gz ]; then
echo "Skull-stripped brain not found, using bet with a fractional intensity threshold of 0.2"
# Note: This fractional intensity appears to work well for most of the subjects in the
# Flanker dataset. You may want to change it if you modify this script for your own study.
bet2 anat/${subj}_T1w.nii.gz \
anat/${subj}_T1w_brain_f02.nii.gz -f 0.2
fi
# Copy the design files into the subject directory, and then
# change “sub-08” to the current subject number
cp ../design_run1.fsf .
cp ../design_run2.fsf .
# 替换*.fsf中的"sub-08"为当前需要处理的被试名称
# Note that we are using the | character to delimit the patterns
# instead of the usual / character because there are / characters
# in the pattern.
sed -i '' "s|sub-08|${subj}|g" \
design_run1.fsf
sed -i '' "s|sub-08|${subj}|g" \
design_run2.fsf
# 直接使用feat执行*.fsf脚本
# Usage: feat <design.fsf>
# Now everything is set up to run feat
echo "===> Starting feat for run 1"
feat design_run1.fsf
echo "===> Starting feat for run 2"
feat design_run2.fsf
echo
# Go back to the directory containing all of the subjects, and repeat the loop
cd ..
done
echo
脚本的开头是一个shebang和一些注释,描述了脚本的具体操作;然后使用反引号来扩展seq -w 1 26,以创建一个循环,在所有被试上运行代码的主体。该脚本使用一个条件来检查是否存在颅骨剥离后的结构像数据,如果不存在,则生成。然后编辑模板design*.fsf文件,将字符串sub-08替换为当前受试者的姓名。*.fsf文件使用命令feat运行,就像从命令行运行FEAT图形用户界面一样。整个脚本中都使用了echo命令,让用户知道何时运行新的步骤。
只需在Flanker目录下输入bash run_1stLevel_Analysis.sh即可运行脚本。当运行一个新步骤时,echo命令将向终端打印文本,HTML页面将跟踪预处理和统计的进度。
该脚本将循环处理Flanker数据集中的所有受试者,并为每次运行做预处理和统计分析。这需要的时间取决于你的机器有多快,但应该需要2-4小时左右。请确保对每个被试进行质量检查,就像在预处理教程中做的那样。